Anti-NFAT1 antibody [25A10.D6.D2] - ChIP Grade
| Name | Anti-NFAT1 antibody [25A10.D6.D2] - ChIP Grade |
|---|---|
| Supplier | Abcam |
| Catalog | ab2722 |
| Prices | $403.00 |
| Sizes | 100 µg |
| Host | Mouse |
| Clonality | Monoclonal |
| Isotype | IgG1 |
| Clone | 25A10.D6.D2 |
| Applications | IHC-P FC EMSA ICC/IF IP WB IHC-F ICC/IF ICC/IF ChIP |
| Species Reactivities | Mouse, Rat, Human |
| Antigen | Synthetic peptide corresponding to Mouse NFAT1 aa 51-69 |
| Description | Mouse Monoclonal |
| Gene | NFATC2 |
| Conjugate | Unconjugated |
| Supplier Page | Shop |
Product images
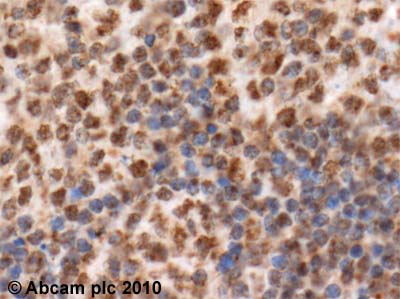 ab2722 (4µg/ml) staining NFAT in human tonsil, using an automated system (DAKO Autostainer Plus). Using this protocol there is strong nuclear and weak cytoplasmic staining.Sections were rehydrated and antigen retrieved with the Dako 3 in 1 AR buffer EDTA pH 9.0 in a DAKO PT link. Slides were peroxidase blocked in 3% H2O2 in methanol for 10 mins. They were then blocked with Dako Protein block for 10 minutes (containing casein 0.25% in PBS) then incubated with primary antibody for 20 min and detected with Dako envision flex amplification kit for 30 minutes. Colorimetric detection was completed with Diaminobenzidine for 5 minutes. Slides were counterstained with Haematoxylin and coverslipped under DePeX. Please note that, for manual staining, optimization of primary antibody concentration and incubation time is recommended. Signal amplification may be required.
ab2722 (4µg/ml) staining NFAT in human tonsil, using an automated system (DAKO Autostainer Plus). Using this protocol there is strong nuclear and weak cytoplasmic staining.Sections were rehydrated and antigen retrieved with the Dako 3 in 1 AR buffer EDTA pH 9.0 in a DAKO PT link. Slides were peroxidase blocked in 3% H2O2 in methanol for 10 mins. They were then blocked with Dako Protein block for 10 minutes (containing casein 0.25% in PBS) then incubated with primary antibody for 20 min and detected with Dako envision flex amplification kit for 30 minutes. Colorimetric detection was completed with Diaminobenzidine for 5 minutes. Slides were counterstained with Haematoxylin and coverslipped under DePeX. Please note that, for manual staining, optimization of primary antibody concentration and incubation time is recommended. Signal amplification may be required.
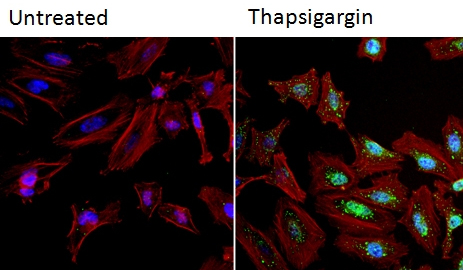 Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) in HeLa cells. Formalin fixed cells were permeabilized with 0.1% Triton X-100 in TBS for 10 minutes at room temperature and blocked with 1% BSA for 15 minutes at room temperature. Cells were left untreated (left panel) or treated with 1uM staurosporine (right panel) for 3 hours and incubated with ab2722 (1:100) for at least 1 hour at room temperature, washed with PBS, and incubated with a DyLight 488 conjugated goat anti-mouse IgG secondary antibody (1:400) for 30 minutes at room temperature. F-Actin (red) was stained with DyLight 554 Phalloidin and nuclei (blue) were stained with Hoechst 33342 dye. Images were taken at 20X magnification.
Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) in HeLa cells. Formalin fixed cells were permeabilized with 0.1% Triton X-100 in TBS for 10 minutes at room temperature and blocked with 1% BSA for 15 minutes at room temperature. Cells were left untreated (left panel) or treated with 1uM staurosporine (right panel) for 3 hours and incubated with ab2722 (1:100) for at least 1 hour at room temperature, washed with PBS, and incubated with a DyLight 488 conjugated goat anti-mouse IgG secondary antibody (1:400) for 30 minutes at room temperature. F-Actin (red) was stained with DyLight 554 Phalloidin and nuclei (blue) were stained with Hoechst 33342 dye. Images were taken at 20X magnification.
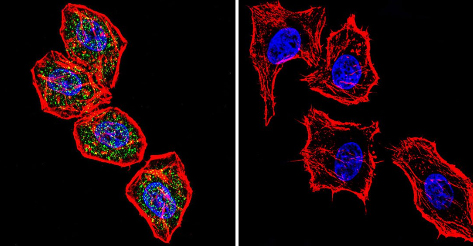 Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in HeLa cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in HeLa cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
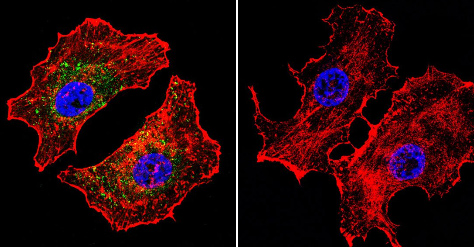 Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in MCF-7 cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in MCF-7 cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
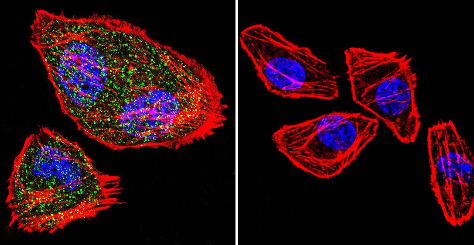 Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in U251 Cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
Immunocytochemistry/Immunofluorescence analysis of NFAT1 (green) shows staining in U251 Cells. F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) is shown. Cells were grown on chamber slides and fixed with formaldehyde prior to staining. Cells were inbuated without (control) or with ab2722 (1:20) over night at 4°C, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody. Images were taken at 60X magnification.
![Anti-NFAT1 antibody [25A10.D6.D2] - ChIP Grade (ab2722) at 1 µg/ml + Spleen (Human) Tissue Lysate - adult normal tissue (ab29699) at 10 µgSecondaryGoat Anti-Mouse IgG H&L (HRP) preadsorbed (ab97040) at 1/5000 dilution ( )developed using the ECL techniquePerformed under reducing conditions.](http://www.bioprodhub.com/system/product_images/ab_products/2/sub_3/29828_NFAT1-Primary-antibodies-ab2722-3.jpg) Anti-NFAT1 antibody [25A10.D6.D2] - ChIP Grade (ab2722) at 1 µg/ml + Spleen (Human) Tissue Lysate - adult normal tissue (ab29699) at 10 µgSecondaryGoat Anti-Mouse IgG H&L (HRP) preadsorbed (ab97040) at 1/5000 dilution ( )developed using the ECL techniquePerformed under reducing conditions.
Anti-NFAT1 antibody [25A10.D6.D2] - ChIP Grade (ab2722) at 1 µg/ml + Spleen (Human) Tissue Lysate - adult normal tissue (ab29699) at 10 µgSecondaryGoat Anti-Mouse IgG H&L (HRP) preadsorbed (ab97040) at 1/5000 dilution ( )developed using the ECL techniquePerformed under reducing conditions.
![Overlay histogram showing Jurkat cells stained with ab2722 (red line). The cells were fixed with 80% methanol (5 min) and then permeabilized with 0.1% PBS-Tween for 20 min. The cells were then incubated in 1x PBS / 10% normal goat serum / 0.3M glycine to block non-specific protein-protein interactions followed by the antibody (ab2722, 2µg/1x106 cells) for 30 min at 22ºC. The secondary antibody used was DyLight® 488 goat anti-mouse IgG (H+L) (ab96879) at 1/500 dilution for 30 min at 22ºC. Isotype control antibody (black line) was mouse IgG1 [ICIGG1] (ab91353, 2µg/1x106 cells) used under the same conditions. Acquisition of >5,000 events was performed.](http://www.bioprodhub.com/system/product_images/ab_products/2/sub_3/29829_NFAT1-Primary-antibodies-ab2722-4.jpg) Overlay histogram showing Jurkat cells stained with ab2722 (red line). The cells were fixed with 80% methanol (5 min) and then permeabilized with 0.1% PBS-Tween for 20 min. The cells were then incubated in 1x PBS / 10% normal goat serum / 0.3M glycine to block non-specific protein-protein interactions followed by the antibody (ab2722, 2µg/1x106 cells) for 30 min at 22ºC. The secondary antibody used was DyLight® 488 goat anti-mouse IgG (H+L) (ab96879) at 1/500 dilution for 30 min at 22ºC. Isotype control antibody (black line) was mouse IgG1 [ICIGG1] (ab91353, 2µg/1x106 cells) used under the same conditions. Acquisition of >5,000 events was performed.
Overlay histogram showing Jurkat cells stained with ab2722 (red line). The cells were fixed with 80% methanol (5 min) and then permeabilized with 0.1% PBS-Tween for 20 min. The cells were then incubated in 1x PBS / 10% normal goat serum / 0.3M glycine to block non-specific protein-protein interactions followed by the antibody (ab2722, 2µg/1x106 cells) for 30 min at 22ºC. The secondary antibody used was DyLight® 488 goat anti-mouse IgG (H+L) (ab96879) at 1/500 dilution for 30 min at 22ºC. Isotype control antibody (black line) was mouse IgG1 [ICIGG1] (ab91353, 2µg/1x106 cells) used under the same conditions. Acquisition of >5,000 events was performed.
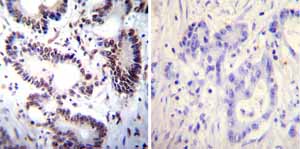 Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human colon carcinoma tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human colon carcinoma tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
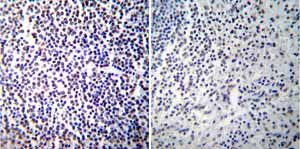 Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human spleen tissue tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human spleen tissue tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
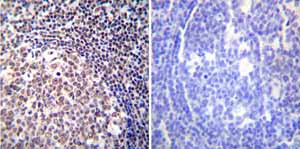 Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human tonsil tissue tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
Immunohistochemistry was performed on both normal and cancer biopsies of deparaffinized Human tonsil tissue tissues. To expose target proteins heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer microwaved for 8-15 minutes. Following antigen retrieval tissues were blocked in 3% BSA-PBS for 30 minutes at room temperature. Tissues were then probed at a dilution of 1:100 with a mouse monoclonal antibody recognizing NFATc2 ab2722 or without primary antibody (negative control) overnight at 4°C in a humidified chamber. Tissues were washed extensively with PBST and endogenous peroxidase activity was quenched with a peroxidase suppressor. Detection was performed using a biotin-conjugated secondary antibody and SA-HRP followed by colorimetric detection using DAB. Tissues were counterstained with hematoxylin and prepped for mounting.
Product References
Bookmarking promoters in mitotic chromatin: poly(ADP-ribose)polymerase-1 as an - Bookmarking promoters in mitotic chromatin: poly(ADP-ribose)polymerase-1 as an
Lodhi N, Kossenkov AV, Tulin AV. Nucleic Acids Res. 2014 Jun;42(11):7028-38.
NFAT1 and NFAT3 cooperate with HDAC4 during regulation of alternative splicing of - NFAT1 and NFAT3 cooperate with HDAC4 during regulation of alternative splicing of
Kosiorek M, Podszywalow-Bartnicka P, Zylinska L, Pikula S. PLoS One. 2014 Jun 6;9(6):e99118.
The ion channel TRPV1 regulates the activation and proinflammatory properties of - The ion channel TRPV1 regulates the activation and proinflammatory properties of
Bertin S, Aoki-Nonaka Y, de Jong PR, Nohara LL, Xu H, Stanwood SR, Srikanth S, Lee J, To K, Abramson L, Yu T, Han T, Touma R, Li X, Gonzalez-Navajas JM, Herdman S, Corr M, Fu G, Dong H, Gwack Y, Franco A, Jefferies WA, Raz E. Nat Immunol. 2014 Nov;15(11):1055-63.
Role of CTLA4 in the proliferation and survival of chronic lymphocytic leukemia. - Role of CTLA4 in the proliferation and survival of chronic lymphocytic leukemia.
Mittal AK, Chaturvedi NK, Rohlfsen RA, Gupta P, Joshi AD, Hegde GV, Bociek RG, Joshi SS. PLoS One. 2013 Aug 1;8(8):e70352.
CD3-T cell receptor co-stimulation through SLAMF3 and SLAMF6 receptors enhances - CD3-T cell receptor co-stimulation through SLAMF3 and SLAMF6 receptors enhances
Chatterjee M, Hedrich CM, Rauen T, Ioannidis C, Terhorst C, Tsokos GC. J Biol Chem. 2012 Nov 2;287(45):38168-77.
Minocycline suppresses activation of nuclear factor of activated T cells 1 - Minocycline suppresses activation of nuclear factor of activated T cells 1
Szeto GL, Pomerantz JL, Graham DR, Clements JE. J Biol Chem. 2011 Apr 1;286(13):11275-82.
TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental - TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental
Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, Raz E. J Clin Invest. 2010 Feb;120(2):570-81.
NIP45 controls the magnitude of the type 2 T helper cell response. - NIP45 controls the magnitude of the type 2 T helper cell response.
Fathman JW, Gurish MF, Hemmers S, Bonham K, Friend DS, Grusby MJ, Glimcher LH, Mowen KA. Proc Natl Acad Sci U S A. 2010 Feb 23;107(8):3663-8. doi:
NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed - NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed
Dammermann W, Zhang B, Nebel M, Cordiglieri C, Odoardi F, Kirchberger T, Kawakami N, Dowden J, Schmid F, Dornmair K, Hohenegger M, Flugel A, Guse AH, Potter BV. Proc Natl Acad Sci U S A. 2009 Jun 30;106(26):10678-83. doi:
Cognitive decline in Alzheimer's disease is associated with selective changes in - Cognitive decline in Alzheimer's disease is associated with selective changes in
Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H 3rd, Kraner SD, Norris CM. J Neurosci. 2009 Oct 14;29(41):12957-69.